
Eroxon® / MED3000 is a topical gel applied directly to the head (glans) of the penis for the treatment of male erectile dysfunction ("ED").
Eroxon® is the agreed brand name in certain regions such as the EU whereas MED3000 continues to be the internal code name used by Futura as well as when referring to countries where regulatory approval or commercial distribution agreements have not yet been achieved.
MED3000
MED3000 is a topical gel applied directly to the head (glans) of the penis for the treatment of male erectile dysfunction ("ED").
MED3000 has been granted marketing authorisation by the FDA in the US, CE marked in Europe, UKCA marked in the UK and is gaining regulatory approval in a growing number of countries across the world. Mexico, Saudi Arabia and Australia are the latest countries to approve MED3000 as a clinically proven topical treatment for adult men with ED that helps men get an erection within 10 minutes.
Launches have commenced in a number of countries in Europe (including the UK) and in the United Arab Emirates by our commercial partners under the brand name Eroxon®. Futura was granted marketing authorisation by the FDA in June 2023 as a De Novo Medical Device with OTC classification. In July 2023 Futura entered into a ground-breaking licensing agreement with Haleon, a world leading consumer healthcare company for exclusive rights to commercialise MED3000 in the USA.
MED3000 is a unique formulation using volatile solvent components specifically tailored for the treatment of ED.
MED3000 is a treatment applied directly to the head (glans) of the penis and massaged for 15 seconds. ED sufferers or their partners can apply the gel directly to the man’s penis as part of foreplay. It is fast-acting helping men get an erection within 10 minutes, and easy to use helping to restore spontaneity and intimacy in the relationship whilst offering an excellent safety profile.
MED3000 Regulatory
Europe
MED3000 is authorised as a Class 2B device in the EU/EEA under the European Medical Device Regulation (MDR), allowing MED3000 to be marketed directly to consumers without the need of a doctor’s prescription across the European Union. MED3000 is the first clinically proven, pan-European topical ED treatment available without a doctor's prescription.
In April 2022, Futura was granted UKCA certification for MED3000, supplementing the EU MDR authorisation. The UKCA mark willreplace the CE mark for medical devices in Great Britain from 1 July 2030.
USA
In June 2023, MED3000 was granted marketing authorisation by the FDA in the USA following a regulatory submission to the FDA for authorisation as a De Novo Medical Device in October 2022, with the potential to be the first major ED treatment available OTC in the USA.
Rest of world
Our commercial partners have filed regulatory submissions in various countries with six regulatory approvals in the Middle East (including Saudi Arabia and the United Arab Emirates), as well as Mexico and Australia, with more to follow.
MED3000 Mode of Action
MED3000 works through a unique evaporative mode of action. MED3000’s combination of volatile components creates a novel action that stimulates nerve sensors in the highly innervated glans penis by a cooling and recovery warming effect, rapidly leading to smooth muscle relaxation, tumescence and erection as shown on the diagram below.
The glans penis is very highly innervated and there are sensors which are reactive to a range of physical sensations, including touch, pressure and temperature. Futura conducted further research and analysis in 2020 which demonstrated the mode of action for MED3000 as shown in the graph.
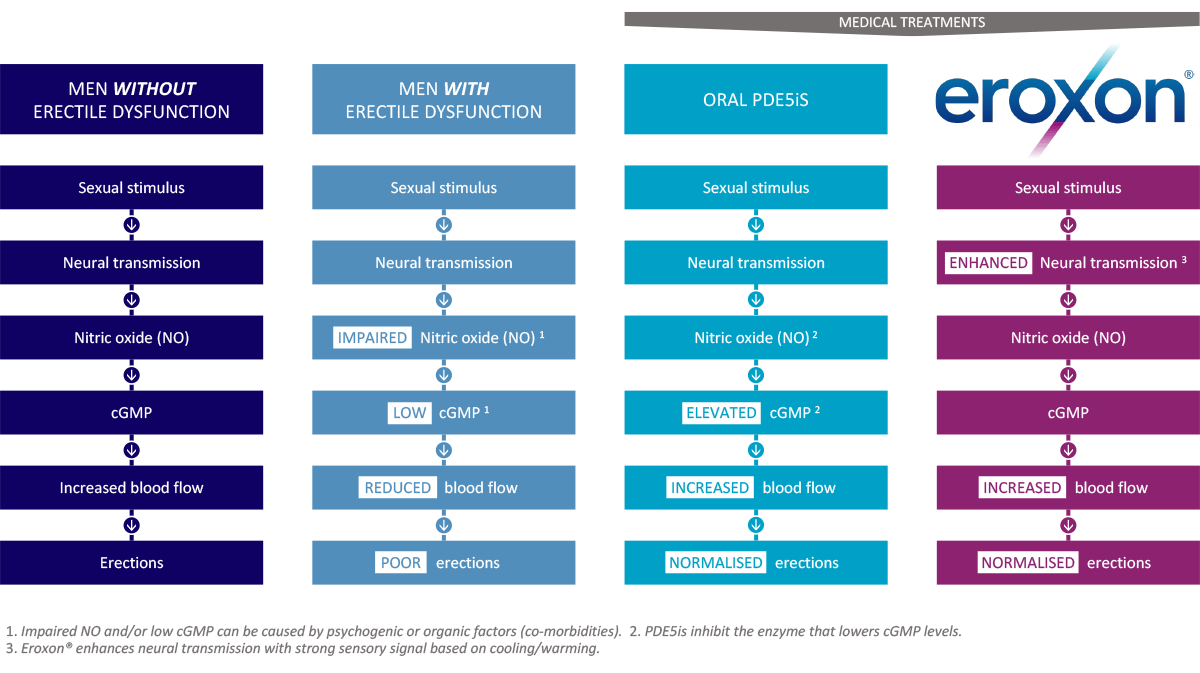
Men without ED
This shows the natural process that would lead to an erection.
Men with ED
This shows what has gone wrong when a man suffers from ED. The production of nitric oxide is impaired leading to low cGMP and therefore reduced blood flow in the penis and poor erections.
Oral medications
This shows how current first-line treatments oral medication work. Oral PDE5i's act at the cGMP levels, elevate those levels in the blood which increases blood flow into the penis and leads to normalised erections.
Eroxon®
This shows how Eroxon® works. Eroxon® acts earlier in the process, at the neural transmitter level, which ultimately increases blood flow to the penis, resulting in normalised erections. Eroxon® kick-starts the normal physiology process that leads to an erection through a unique temperature change effect.
Clinical Data
MED3000 is supported by efficacy and safety data from a range of studies including the completed Phase 3 studies FM57 and a US confirmatory Phase 3 study, FM71.
FM57 Phase 3 Study
The 1,000 patient study included approximately 60 centres across nine Central and Eastern European countries. FM57 was a randomised, double-blind, placebo-controlled, home use, parallel group clinical trial. Subjects being enrolled into FM57 for the initial four weeks had to attempt intercourse on at least four occasions in order to establish the severity of their ED, known as the pre-treatment ‘baseline’.
FM57, the Phase 3 study was designed to investigate the efficacy and safety of a range of topically applied gels using IIEF-EF and SEP 2 and 3 as co-primary clinical endpoints in mild, moderate and severe ED patients.
FM57 Clinical Results
In study FM57, MED3000 consistently showed statistical improvement over pre-treatment, baseline and achieved clinically important differences across all ED severities. MED3000 also showed efficacy and a rapid speed of onset with an excellent safety profile.
FM57 demonstrated that MED3000 has the potential to be a highly effective, clinically proven, topical treatment for erectile dysfunction. MED3000 has a unique evaporative mode of action which the Company believes stimulates nerve endings in the glans penis by a cooling and recovery warming effect to cause an erection.
MED3000 results demonstrated a highly statistically significant improvement (p<0.001) in erectile function across ‘pooled’ patient severities (mild, moderate, and severe) as well as being statistically significantly superior within the separate mild, moderate and severe groups, compared to before treatment baseline, along with an excellent safety profile.
Data analysed was positive on measures of clinically meaningful benefit which physicians, ED sufferers and regulators view as increasingly important. MED3000 had a significant clinically meaningful effect in over 60% of patients as calculated using the Rosen1 and Araujo2 statistical methods, standard assessment techniques for measuring Patient Reported Outcomes recognised and accepted by leading ED experts.
MED3000 begins to work immediately in some patients, with over 60% of patients seeing onset of their erection within 10 minutes of application, substantially faster than reported for PDE5i's (oral tablets) with significant benefits for spontaneous rather than pre-planned sexual intercourse.
Safety and tolerability data were also highly positive, with no serious adverse events recorded in any patient, or their female partners.
- Rosen at al. "Minimal Clinically Important Differences in the Erectile Function Domain of the International Index of Erectile Function Scale", European Eurology, July 2011;
- Araujo et al. "Minimal Clinically Important Differences in the Vaginal Insertionand Successful Intercourse Items of the Sexual Encounter Profile" Journal of Sexual Medicine, January 2012
FM57 STUDY DESIGN
- FM57 was a multicentre, randomised, double blind, placebo controlled, home use, parallel group clinical trial of a range of topically applied gels conducted across nine Central and Eastern European countries.
- 1,000 subjects with mild, moderate or severe ED comparing the efficacy of a range of topically applied gels including “MED3000” using IIEF-EF and SEP 2 and 3 as co-primary clinical endpoints.
- Subjects being enrolled into the FM57 study had to attempt intercourse on at least four occasions in the initial four weeks in order to establish the severity of their ED before treatment known as the ‘baseline’.
FM57 CONCLUSIONS
- MED3000 consistently showed statistical improvements against before treatment (baseline) with clinically important differences across all ED severities.
- MED3000 also showed statistical improvements on both GAQ (Global Assessment Questionnaire) and SEAR (Self-Esteem And Relationship Questionnaire) secondary endpoints.
- MED3000 also showed meaningful clinical differences across all ED patient groups, mild, moderate and severe, which is a key evaluation criteria for regulators.
- MED3000 had an excellent safety profile with very low incidence of adverse events in males and female partners.
- Results demonstrated that MED3000 has the potential to be an effective, clinically proven, topical treatment for erectile dysfunction.
FM71 - US Confirmatory Phase 3 Study
FM71 was a multi-centre, randomised, open-label, home use, parallel group, clinical investigation of topically-applied MED3000 gel compared to a well-known US prescription oral medication for the treatment of ED in 96 subjects over a 24-week period. The trial design and clinical endpoints were agreed with the FDA as a confirmatory clinical trial for the US regulatory dossier which led to the FDA approval of MED3000.
FM71 investigated the efficacy and safety of MED3000 gel in male subjects clinically diagnosed with mild, moderate or severe ED against baseline (pre-treatment). Subjects were recruited from the United States (African Americans), Poland, Georgia and Bulgaria and included men who had organic and psychological ED or a combination of both.
Subjects enrolled into the FM71 study for the initial four weeks had to attempt intercourse on at least four occasions in order to establish the severity of their ED known as the ‘baseline’, after which MED3000 was used as per trial protocol for 24 weeks.
A well-known US prescription oral medication was included in a comparator arm for informational purposes only, to assess relative safety, speed of onset and overall efficacy. This enabled the FDA to determine the relative benefit/risk ratio of MED3000 versus a commercially available prescription comparator product.
FM71 Clinical Results
FM71 results were highly positive, meeting all primary and secondary endpoints, were in-line with data generated in the previous Phase 3 clinical study (“FM57”) and are broadly comparable with results from a recent “real world”, home user study conducted by our commercial partner, enabling FDA submission of MED3000 as De Novo Medical Device in October 2022 with FDA granting marketing authorisation in June 2023.
Data demonstrates that MED3000 presents an effective clinically proven treatment for erectile dysfunction with a rapid speed of onset and a favourable benefit versus risk profile ideally suited for over the counter classification.
It will provide an alternative to existing ED treatments, that require a doctor’s prescription, and for those ED sufferers seeking fewer systemic side-effects, and a spontaneous intercourse experience. It also provides an important treatment option for those ED sufferers who are currently precluded from using current prescription treatments such as men taking nitrate medication.
FM57 STUDY DESIGN
- FM71 is a multi-centre, randomised, open-label, home use, parallel group, clinical investigation of topically-applied MED3000 gel compared to a well-known US prescription oral medication for the treatment of ED over a 24-week period.
- The trial design and clinical endpoints were agreed with the FDA as a confirmatory clinical trial for the US regulatory dossier for MED3000 which we continue to target filing by the end of September 2022.
- FM71 investigated the efficacy and safety of MED3000 gel in 96 male subjects clinically diagnosed with a mix of mild, moderate and severe ED against baseline (pre-treatment). Subjects were recruited from the United States (African Americans), Poland, Georgia and Bulgaria and included men who had organic and psychological ED or a combination of both.
- Subjects enrolled into the FM71 study for the initial four weeks had to attempt intercourse on at least four occasions in order to establish the severity of their ED known as the ‘baseline’, after which MED3000 was used as per trial protocol for 24- weeks.
FM57 CONCLUSIONS
- MED3000 demonstrated a highly statistically significant improvement against baseline at 24 weeks in erectile function (as measured by the gold standard, internationally recognised IIEF-EF score) across ‘pooled’ severities of ED (mild, moderate and severe). (Primary Endpoint)The 24-week time point demonstrated durability of response to treatment beyond 12 weeks (studied previously in FM57) which was specifically requested by the FDA.
- MED3000 achieved a clinically important improvement in subjects' erectile function at all time points and was clinically effective in mild, moderate and severe ED sufferers - subjects experienced a 5.73 unit change in IIEF-EF score versus baseline at 24 weeks exceeding the 4-unit difference agreed with the FDA and defined as the Minimal Clinical Important Difference (“MCID”) - (Primary Endpoint)
- MED3000 demonstrated a rapid speed of onset, where subjects noticed an erection at 10-minutes, (Secondary Endpoint) which was demonstrably faster than the well-known prescription oral medication used in the study
- Safety and tolerability data were also highly positive, with no serious adverse events recorded in any patients on MED3000 and overall, with a highly favourable side-effect profile.
Download the poster presented at the 2023 Conference from the European Society for Sexual Medicine
David Ralph et al “MED3000, a clinically proven, fast-acting topical product for Erectile Dysfunction with the prospect of being the first globally available OTC treatment for ED” (MED3000 is the codename used when Eroxon® was in the research stage).
Eroxon® is the agreed brand name in certain regions such as the EU whereas MED3000 continues to be the internal code name used by Futura as well as when referring to countries where regulatory approval or commercial distribution agreements have not yet been achieved.
Click here to go to our product website eroxon.com
Click here to go to our commercial partner website www.eroxon.eu